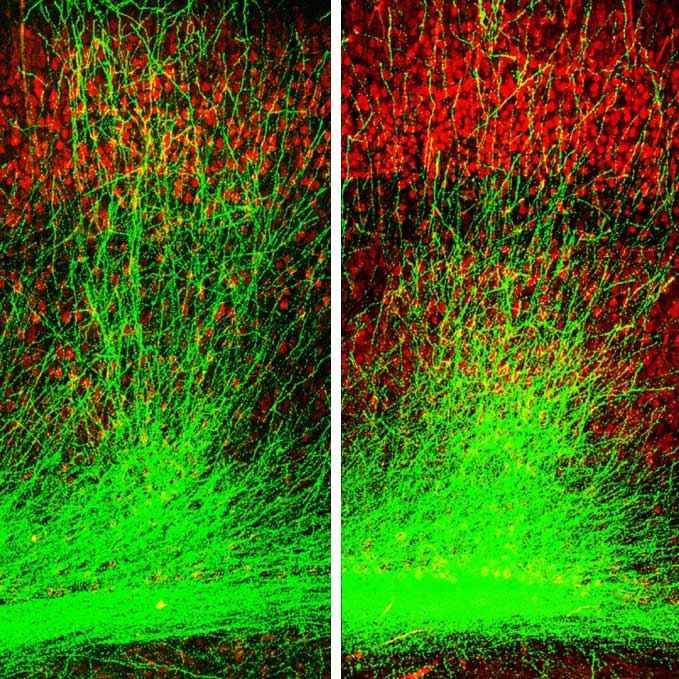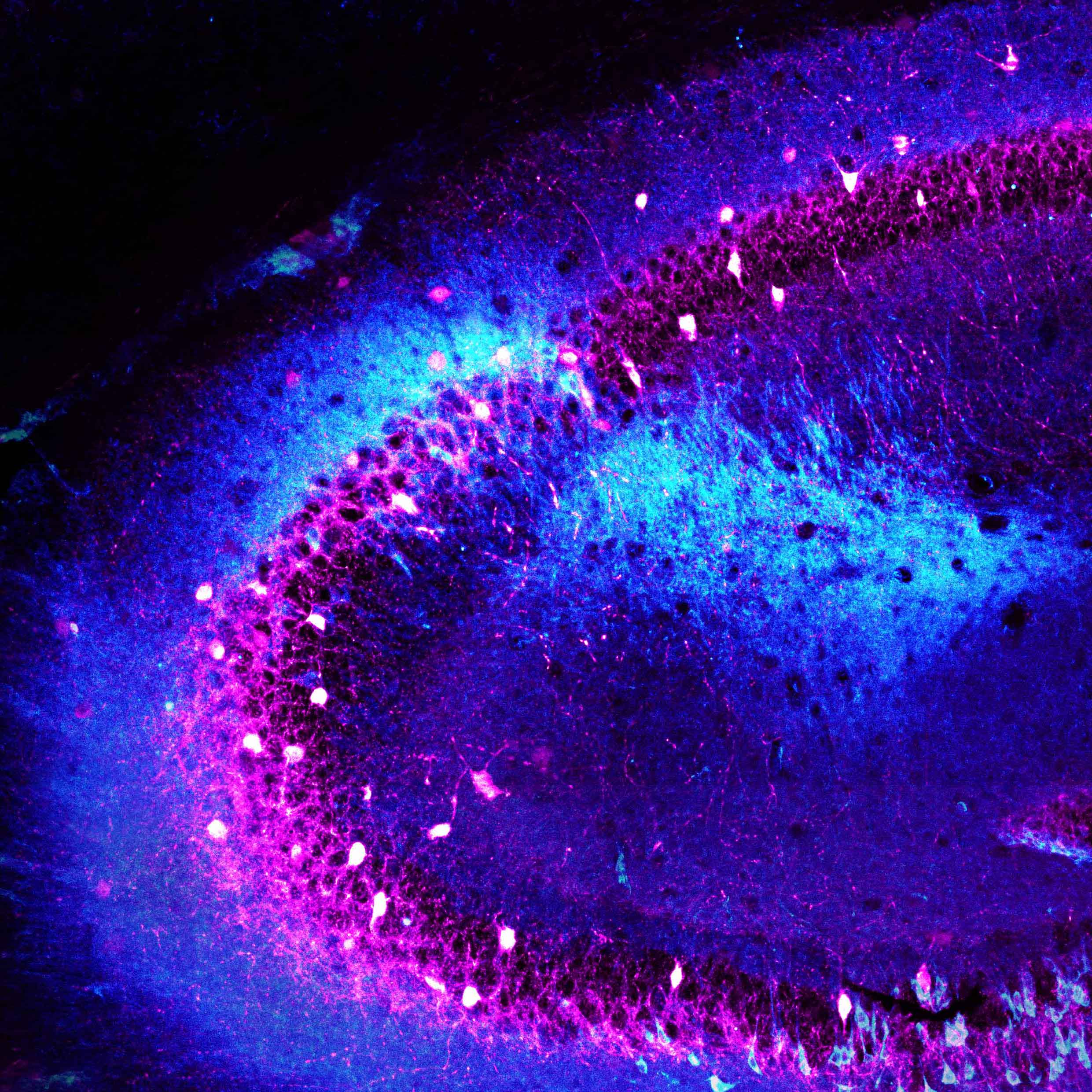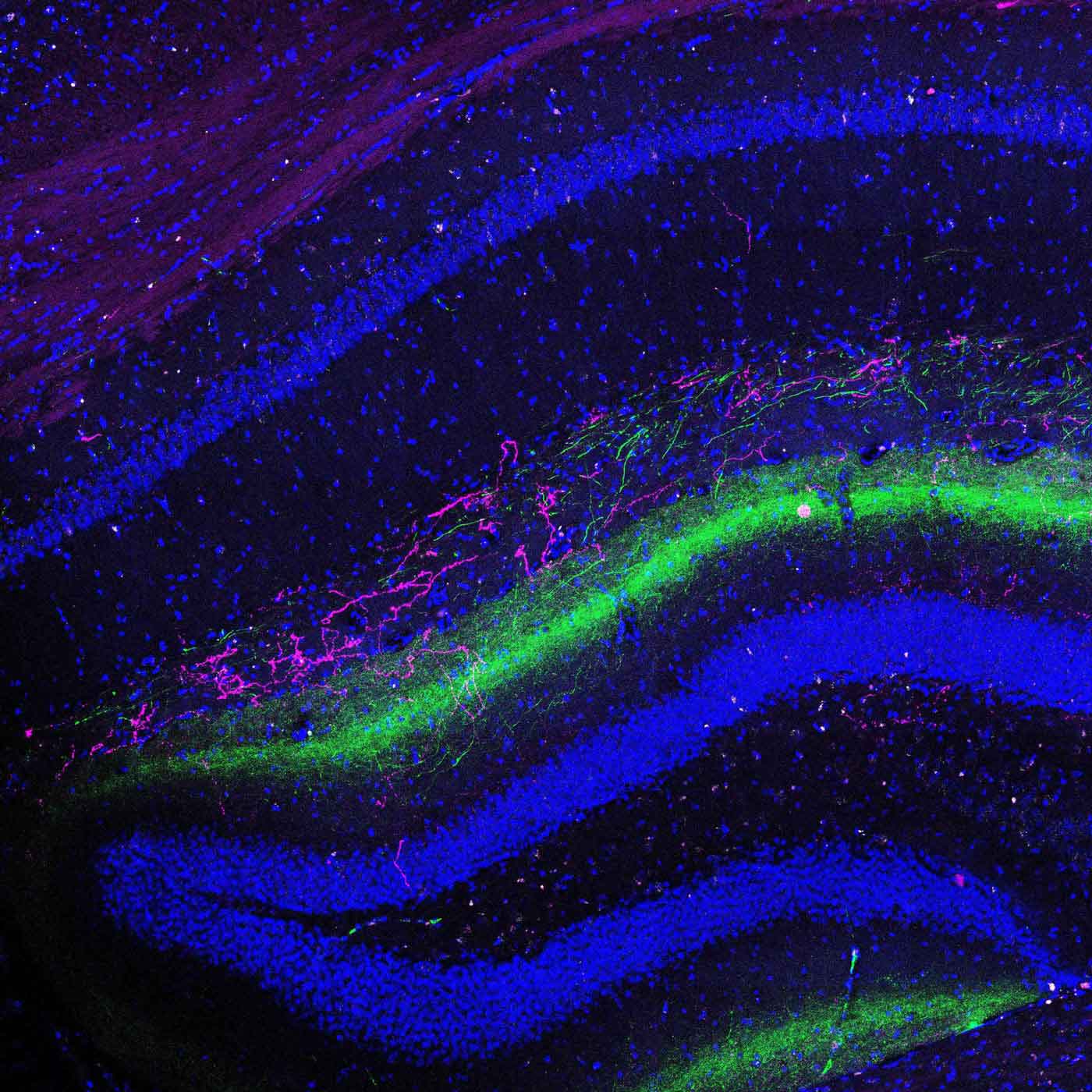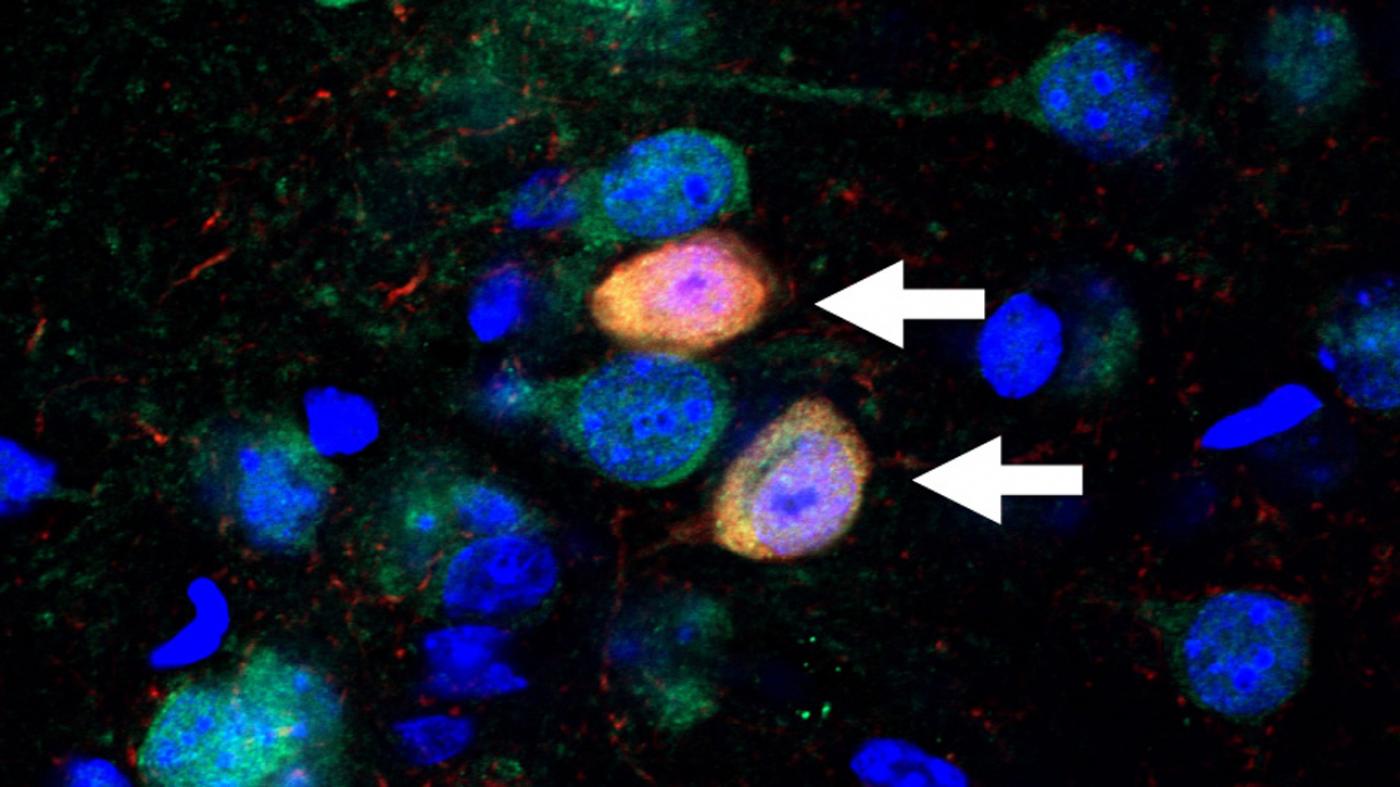
Schizophrenia affects millions of people worldwide, and yet we understand so little about the disease. Joseph Gogos, MD, PhD, a neuroscientist at Columbia’s Mortimer B. Zuckerman Mind Brain Behavior Institute and a professor at Columbia University Medical Center (CUMC) studies schizophrenia’s biological origins. Dr. Gogos has co-authored a new paper, published today in Cell Reports, that sheds light on how a genetic change alters healthy brain function in a mouse model of schizophrenia.
We spoke with Dr. Gogos about what his latest research has revealed, what it could mean for the many people suffering from the disease, and whether it could point to new pharmacological treatments.
What questions were you trying to answer with this study?
It is well known that a deletion to a particular portion of a human chromosome — an area known as 22q11.2 — results in a one-in-three chance of developing schizophrenia. By comparison, the average risk of schizophrenia for those without this mutation is about one in 100. My colleagues and I have been trying to understand what causes this dramatic increase in someone’s risk for developing schizophrenia by focusing on the genes that are removed with the 22q11.2 deletion. One such gene, called PRODH, makes an enzyme whose job is to flush out excess L-proline (an amino acid) from the brain. If PRODH can’t produce the enzyme, L-proline can build up to toxic levels. We’ve known for many years now that elevated L-proline levels results in increased risk of psychosis — including schizophrenia — but how exactly this happens remained a mystery.
How did you go about finding answers?
About five years ago we noticed that L-proline has a similar shape to GABA, another molecule found in the brain. GABA serves as the brain’s main inhibitory neurotransmitter, a chemical that keeps brain activity levels in check. The striking resemblance of L-proline to GABA made us wonder whether elevated L-proline levels might cause dysfunction by disrupting GABA activity. To pursue this observation further, we looked at whether L-proline was somehow interacting with GABA receptors, the molecules that normally receive GABA signals.
What did you find?
In experiments with laboratory mice, we found that at levels of L-proline one would find in a brain — even one affected by schizophrenia — L-proline didn’t seem to have an effect on GABA receptors. So we wondered whether there would be a less obvious way that L-proline and GABA were linked.
Indeed, our experiments revealed a kind of molecular confusion. Because L-proline structurally resembles GABA, the protein that normally makes GABA, known as glutamate decarboxylase, reacts as if L-proline were GABA, and slows down GABA production. The result is a nervous system that acts like a car with worn-out brakes.
What’s the connection between problems with GABA and psychiatric disorders such as schizophrenia?
Decades of research has shown that keeping brain activity in sync, through waves called gamma oscillations, is very important to a healthy mind. Disrupting them contributes to some of schizophrenia’s key symptoms, such as memory and attention impairments. Importantly, to function properly, gamma oscillations rely on GABA.
In today’s study, in collaboration with my CUMC colleague Dr. Joshua Gordon, we found that gamma oscillations were profoundly disrupted in mice with mutations in the PRODH gene. Those mutations increase L-proline concentrations and slow-down GABA production. This exciting finding helped us to draw a direct line between elevated L-proline levels and disruptions in higher brain function, such as those observed in schizophrenia.
Was anything particularly surprising?
What most surprised us was the way in which elevated L-proline affected gamma oscillations. We found the effect of L-proline not where you’d think — the GABA receptors — but rather at a much less obvious place, the protein that makes GABA.
What does this study teach us about brain function more generally?
Despite the fact that our original scope and motivation (the 22q11.2 deletion) was specific, there is a much broader importance of our findings. Recently in biology, there’s been an explosion in “metabolomics,” the study of the small molecules in the human body and brain. There are well over 40,000 structurally different small molecules in the human body, many of which, while not normally used for signaling in the brain, may in fact be capable of altering brain function. So it’s very plausible that disturbances in other small molecules, similar to the case here with L-proline, play an important role in human psychiatric disease. We have already uncovered and discuss additional examples in our paper.
Might this study lead to applications in the treatment of psychiatric disease?
This is a major goal of our work. The deficiencies in GABA production and function that we observe here point to very clear strategies for a pharmacological intervention. If we can employ clinically available “pro-GABA-ergics,” drugs that boost GABA, this could help ameliorate dysfunction in individuals who carry the 22q11.2 deletions. In fact, our preliminary tests of two different FDA-approved pro-GABA-ergic drugs in cells and in mice revealed that these drugs could substantially restore normal function. The fact that these drugs are already FDA-approved will make it simpler to test them in patients — an approach we call “precision-psychiatry.”
What are the next questions that need to be answered?
We want to find out if pro-GABA-ergic therapies correct dysfunction at the behavioral level to actually mitigate symptoms of schizophrenia. Also, our discovery sheds light on how mutations that increase one’s risk of schizophrenia affect the GABA machinery that normally keeps brain activity levels in check. This is just one of many exciting things we’re investigating along these lines. As we continue to get more insights into schizophrenia’s underlying mechanisms, we can begin testing ways to manipulate the GABA machinery for therapeutic purposes.
###
The paper is titled: “Cytosolic accumulation of L-proline disrupts GABA-ergic transmission through GAD blockade.” Additional coauthors include first author Gregg Crabtree, PhD, Alan Park and Joshua Gordon, MD, PhD.