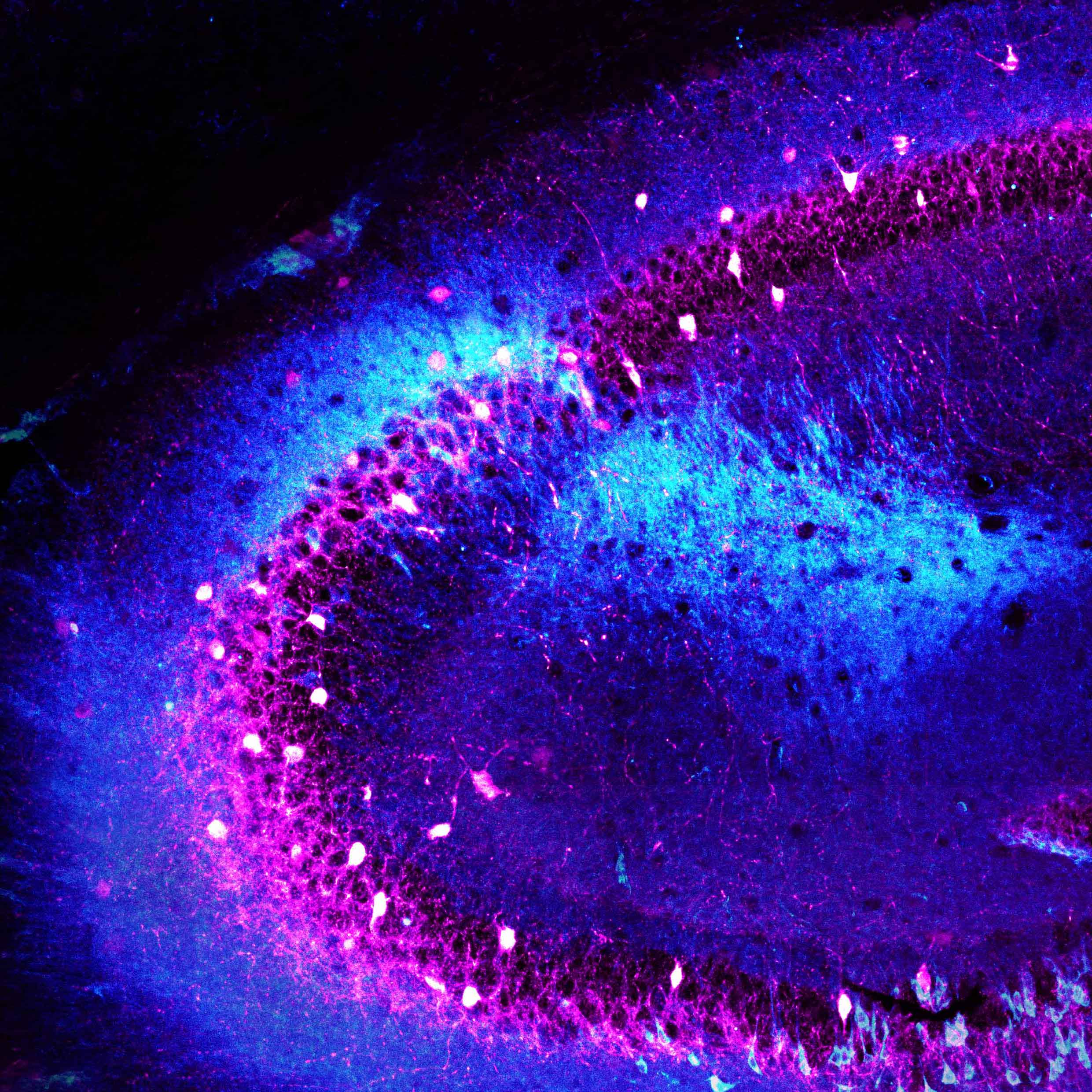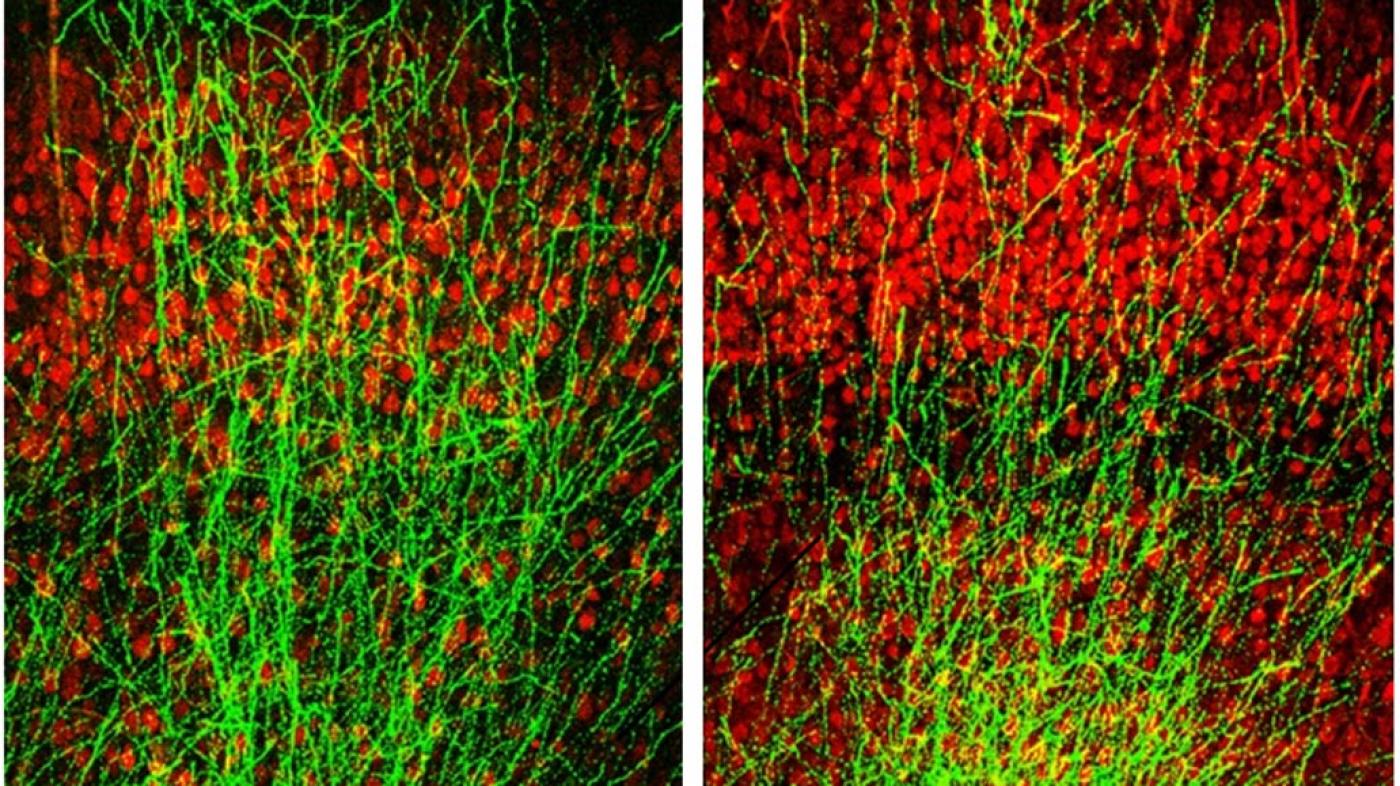
Researchers have successfully disrupted a genetic chain of events in a mouse model of schizophrenia and reversed memory deficits, one of the disorder’s most difficult-to-treat symptoms. This discovery — which builds upon decades of early-stage research — could lead to more effective therapies for the cognitive symptoms of schizophrenia, a psychiatric disorder that affects more than 21 million people worldwide.
In a paper published today in the journal Neuron, scientists at Columbia’s Mortimer B. Zuckerman Mind Brain Behavior Institute, Columbia University Medical Center (CUMC) as well as the New York State Psychiatric Institute (NYSPI) used a chemical compound to regrow connections between brain cells, or neurons, which in turn restored memory deficits. The abnormal or stunted growth of neurons in the brain’s memory centers is a key indicator of schizophrenia.
Understanding how schizophrenia originates in our model lends critical insight into the disorder as a whole, paving the way for improved treatment options that have thus far remained elusive.
The study was performed in mice with a specific genetic mutation known as the 22q11.2 microdeletion. This mutation, which occurs in one percent of people with schizophrenia, is the single largest genetic risk factor for the disease.
“With these findings, we showed that restoring cellular connections reversed memory deficits — a symptom of schizophrenia for which there is no effective treatment,” said Joseph Gogos, MD, PhD, a principal investigator at the Zuckerman Institute, professor of physiology and neuroscience at CUMC and a senior author of the paper. “This represents an invaluable new strategy for treating schizophrenia and highlights the critical importance of basic biological research in psychiatric disorders. Understanding how schizophrenia originates in our model lends critical insight into the disorder as a whole, paving the way for improved treatment options that have thus far remained elusive.”
Paranoia, auditory hallucinations and delusions are some of schizophrenia’s most well known symptoms and can often be controlled with antipsychotic medication. However, other symptoms — like severe disruptions to short-term and verbal memory, reduced attention and a decrease in IQ — have remained largely untreatable. Memory deficits can interfere with an individual’s ability to maintain relationships or a job — essentially cutting them off from the world around them.
“Memory deficits are now considered a core feature of schizophrenia and are believed to be strongly tied to the disorder’s underlying biology,” said Joshua Gordon, MD, PhD, associate professor of psychiatry at CUMC, a research scientist at NYSPI and a senior author of the paper. “Until we can shed light on that biology, these symptoms remain virtually impossible to treat.”
To address this, Dr. Gogos has spent nearly 20 years working to uncover the biological basis of schizophrenia. He and his collaborators developed a laboratory mouse model that exhibited the 22q11.2 microdeletion.
“Schizophrenia affects about one in every 100 people, but for those with a 22q11.2 microdeletion, that chance jumps to one in three,” said Dr. Gogos. “22q11.2 microdeletions remain the single greatest genetic risk factor for developing schizophrenia. This is why our model has proved invaluable in allowing us to trace schizophrenia back to its beginnings.”
In a study published last year, Drs. Gogos, Gordon and their research team revealed how the 22q11.2 microdeletion leads to weaker connections between neurons in key brain regions, resulting in schizophrenia’s memory deficits.
“Normally, neurons grow long branches that connect across long distances, forming tightly interconnected circuits,” said Dr. Gordon. “However, in our mouse models, these branches were stunted in the hippocampus and prefrontal cortex — two regions crucial for memory. Further experiments revealed why: a protein called Gsk3β was clogging the brain.”
Last year’s study showed that the 22q11.2 microdeletion kickstarts a chain reaction that boosts Gsk3β activity right from birth. Overactive Gsk3β stunts the growth and development of neurons in the brain’s memory centers. Over time, this hampers their ability to form strong connections.
In the new study, the researchers tested whether suppressing Gsk3β activity could reverse the damage that ultimately leads to schizophrenia’s memory deficits.
In a series of experiments, the researchers treated their mouse models at a young age (between seven and 28 days after birth), with a chemical compound that blocked Gsk3β activity. Almost immediately, they began to see a difference. As Gsk3β activity dropped, the neurons branched out and formed connections with their neighbors. Communication between brain regions was restored, and — importantly — the mice showed no memory deficits.
“Our work suggests that Gsk3β could be considered a potential target for treating some symptoms of schizophrenia — though there are limitations,” said Dr. Gordon. “For example, whether the same treatment might work in other models of schizophrenia that do not have the 22q11.2 microdeletion. Also, we treated the mice at a young age, but schizophrenia normally presents in adolescence and young adulthood.”
“We’re currently planning to test whether blocking Gsk3β during that equivalent age in our models would also have the same positive effect,” added Dr. Gogos.
“Schizophrenia is a complex disorder — there is no single cause — but disrupted communication between the hippocampus and prefrontal cortex may be a common thread running through many of them,” said Vikaas Sohal, MD, PhD, assistant professor of psychiatry at the University of California, San Francisco School of Medicine, who was not involved with the study. “The findings presented here by Drs. Gogos, Gordon and their team could lead to additional ways of restoring normal communication between these two brain regions — and thus provide effective treatments for schizophrenia that arise from a variety of causes.”
###
This paper is titled: “Developmental inhibition of Gsk3β rescues behavioral and neurophysiological deficits in a mouse model of schizophrenia disposition.” Additional contributors include Makoto Tamura, PhD, the paper’s first author, and Jun Mukai, MD, PhD.
This research was supported by a grant from the National Institute of Mental Health (MH096274). Makoto Tamura is an employee of Mitsubishi Tanabe Pharma Corporation. The authors report no financial or other conflicts of interest.