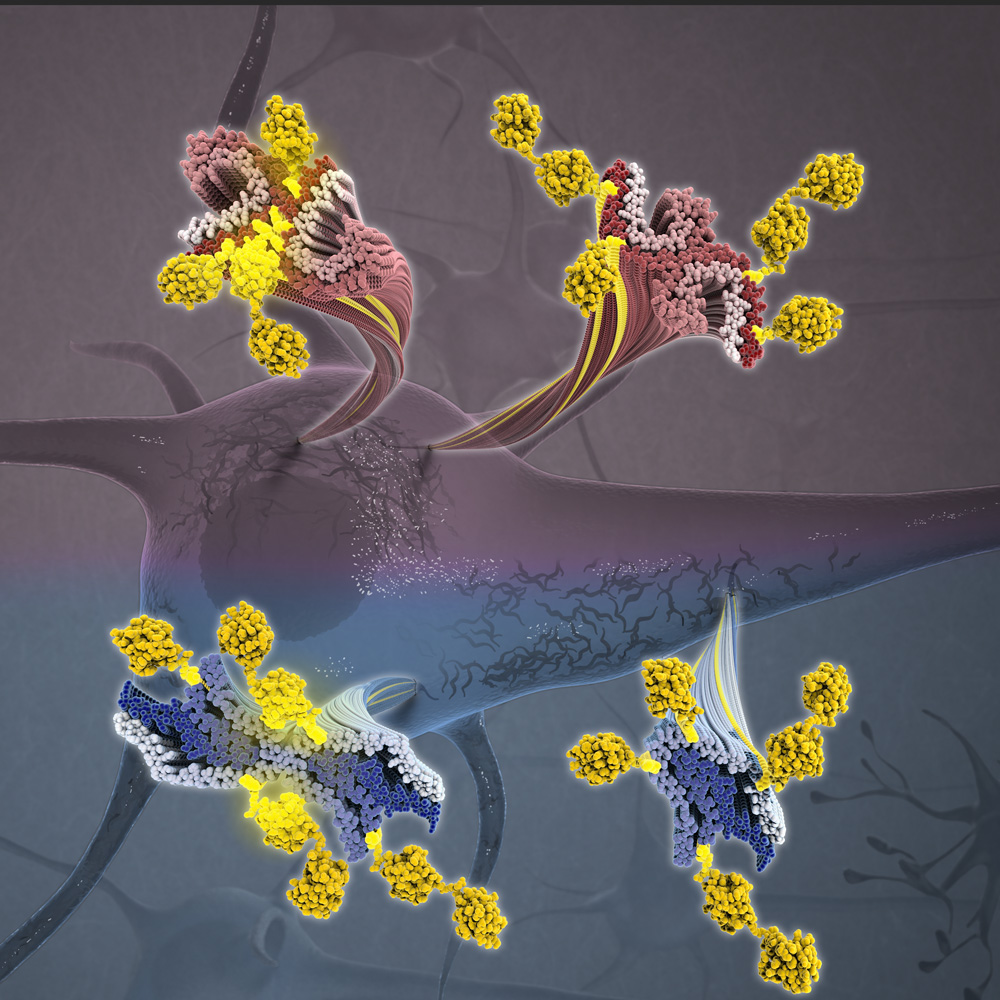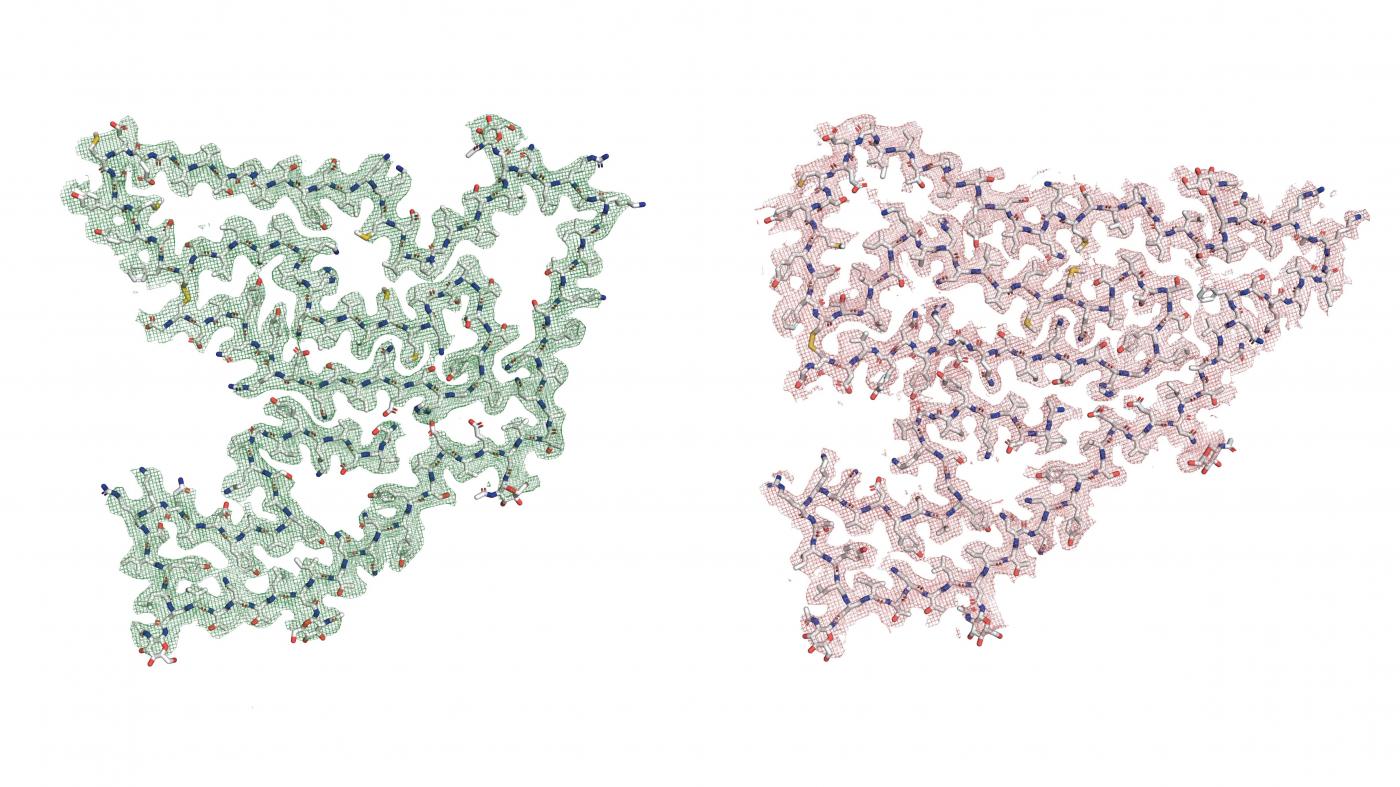
NEW YORK — Take a cell-deep tour of a brain afflicted with Alzheimer’s disease, and you will find minuscule clumps of protein that seem suspicious. Ever since the 1980s, when neuroscientists began identifying these protein tangles, researchers have discovered that other brain diseases have their own tangled-protein signatures.
“Each of these diseases has a unique protein tangle, or fibril, associated with it,” said Anthony Fitzpatrick, PhD, principal investigator at Columbia’s Zuckerman Institute. “These proteins associated with diseases have their own shapes and behaviors,” added Dr. Fitzpatrick, also an assistant professor of biochemistry and molecular biophysics at Columbia University Irving Medical Center and a member of Columbia’s Taub Institute for Research on Alzheimer’s Disease and the Aging Brain.
Published today in Cell, the research by Dr. Fitzpatrick and an international team of 22 collaborators reveals a new fibril in diseased brains, one formed by a protein normally busy cleaning cells.
“We have a surprising and provocative result that we hope could have some bearing on managing neurodegenerative diseases,” said undergraduate Andrew Chang, a co-first author on the paper in the Fitzpatrick lab. Drug researchers have long pursued the tangle-forming proteins as targets for new medicines, but this pursuit so far has largely delivered disappointing results.
Fibril-associated diseases, some common and some rare, collectively affect millions of people around the world. Their incidence is slated to increase as the population grows and people live longer. Untangling what is going on in these neurodegenerative diseases has a personal facet for Dr. Fitzpatrick: He lost an uncle to one of them, progressive supranuclear palsy (PSP).
“We have found that a protein called TMEM106B can form fibrils, and this behavior was not known before,” said Xinyu Xiang, formerly a member of the Fitzpatrick lab at the Zuckerman Institute and now a graduate student at Stanford University’s Department of Structural Biology. “This protein is a core component of lysosomes and endosomes, which are organelles that clean up the junk that builds up in our cells as we get older.”
We have found that a protein called TMEM106B can form fibrils, and this behavior was not known before
Normally, TMEM106B molecules span the membranes of those waste-management organelles. In a feat of laboratory sleuthing, Fitzpatrick’s team discovered that TMEM106B molecules can split into two fragments. Fragments inside the organelles can then self-assemble into what the researchers suspect could be cell-hobbling fibrils.
To make this discovery, the researchers first extracted proteins from brain tissue donated by 11 patients who had died from three neurodegenerative diseases associated with misfolded proteins: PSP, dementia with Lewy bodies (DLB) and frontotemporal lobar degeneration (FTLD). FTLD is the most prevalent form of dementia for those under 60 years of age.
“It’s so motivating to remember that the only way we can do this research is because of people who generously donated their brains,” said Marija Simjanoska, a co-first author and one of the three undergraduates working on the project.
Co-corresponding author Ian Mackenzie, MD, of the University of British Columbia, and co-authors Dennis Dickson, MD, and Leonard Pertrocelli, PhD, of the Mayo Clinic in Florida, helped procure this precious research resource. Joining Drs. Fitzpatrick and Mackenzie as co-corresponding authors on the paper is Michael Stowell, PhD, of the University of Colorado, Boulder. Filling out the 23-member team are researchers from several other institutions, including three in Belgium.
With a world-class cryogenic electron microscope (cryo-EM), the team took snapshots of individual protein molecules at many different angles. From these, the researchers constructed three-dimensional models of the protein in atomic detail. Those models, in turn, helped the researchers identify TMEM106B by making educated guesses about the exact sequence of the protein’s amino-acid building blocks. Much in the way letters string into words with specific meanings, different amino-acid molecules build into proteins, each with its own shape and function.
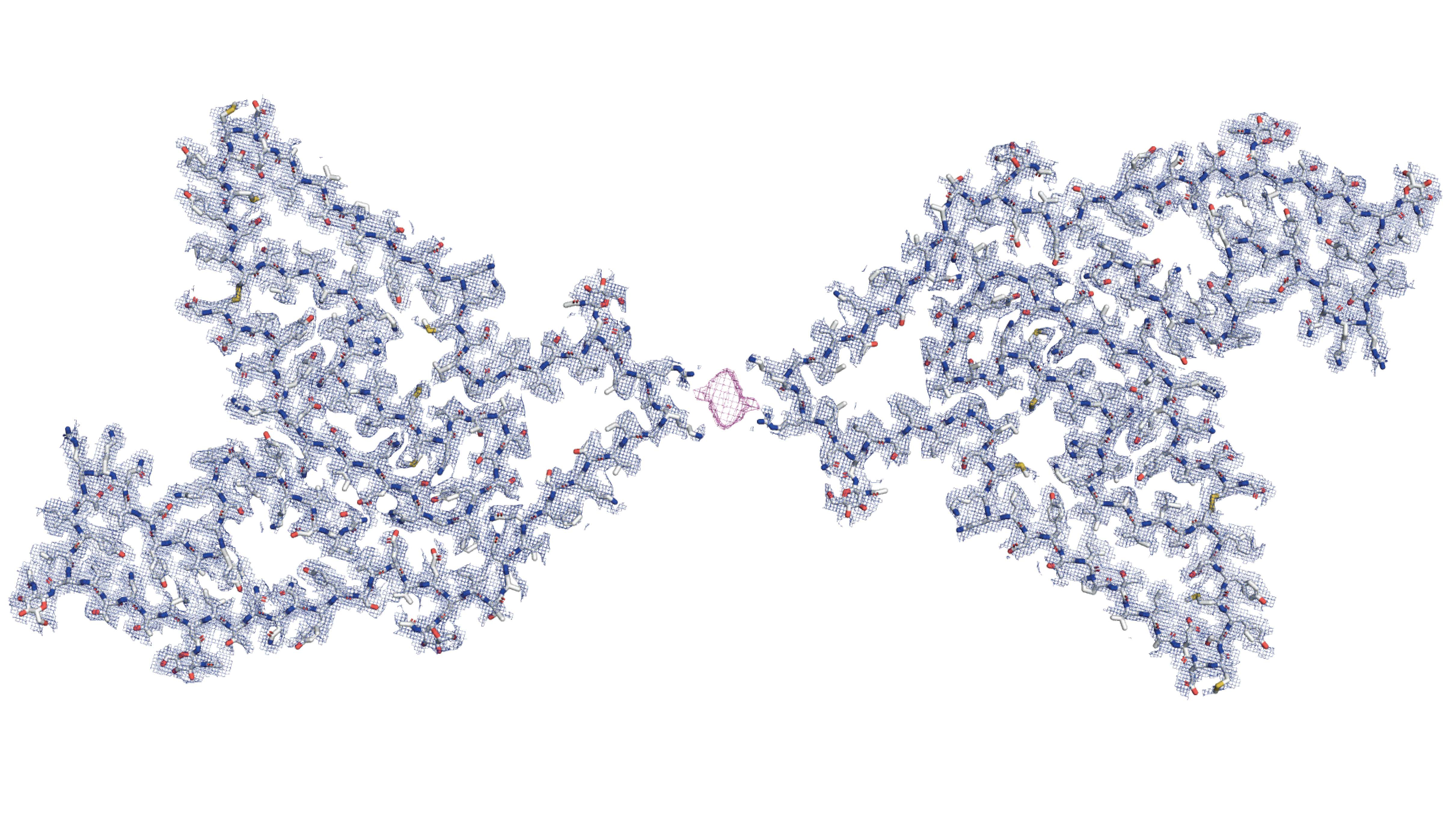
Two fragments of the protein TMEM106B (blue) form a doublet by binding to a currently unidentified non-proteinaceous density (purple mesh).
(Credit: Andrew Chang and Anthony Fitzpatrick / Columbia University’s Zuckerman Institute / Cell)
The researchers fully expected that one of the long-known fibril-forming proteins, such as the tau protein in Alzheimer’s disease, would end up matching with the models from the cryo-EM data. Instead, the matching exercise, which involved searching in a massive database of protein sequences, delivered a head-turning result.
The researchers found that the mysterious protein matched a 135-amino-acid fragment of TMEM106B. That was an exciting revelation because this same protein was identified more than a decade ago in a broad hunt for genes potentially associated with FTLD.
So far, the data in hand shows only that TMEM106B fibrils are present in diseased brain tissue, not that the fibrils cause the diseases. Still, Dr. Fitzpatrick points out, the prevalence of TMEM106B fibrils in tissue from different brain diseases, combined with the protein’s normal place in lysosomes and endosomes, points toward a possible disease-causing role.
In their Cell paper, the researchers speculate that the formation of TMEM106B fibrils disrupts lysosome function, which, in turn, promotes the formation of fibrils made of the other known fibril-forming proteins. These malfunctions could kill brain cells, leading to dementia, movement problems, speech pathologies and other symptoms of Alzheimer’s, PSP, FTLD and other brain diseases with telltale protein tangles.
“We now have a promising new lead,” said Dr. Fitzpatrick. “It could point towards a common thread linking a range of neurodegenerative diseases and could open the way to new interventions.”
###
The Cell paper is titled, “Homotypic fibrillization of TMEM106B across diverse neurodegenerative diseases”
The paper’s authors are Andrew Chang, Xinyu Xiang, Jing Wang, Carolyn Lee, Tamta Arakhamia, Mrija Simjanoska, Chi Wang, Yari Carlomagno, Guoan Zhang, Shikhar Dhingra, Manon Thierry, Jolien Perneel, Bavo Heeman, Lauren M. Forgrave, Michael DeTure, Mari L. DeMarco, Casey N. Cook, Rosa Rademakers, Dennis Dickson, Leonard Petrucelli, Michael H. B. Stowell, and Ian R. A. Mackenzie and Anthony Fitzpatrick.
The authors declare no competing interests.
This work was supported by the National Institutes of Health (NIH)/National Institute of Neurological Disorder and Stroke (UO1NS110438, U54NS110435); the Association for Frontotemporal Degeneration; Canadian Institutes of Health Research (74580); and MCDB Neurodegenerative Disease Fund.